


-
摘要: 采用改进的Humer法合成了石墨烯氧化物,利用搭建的时间分辨光谱探测系统详细探究了Fe3+(浓度为0.5、1、2 mmol/L)对石墨烯氧化物荧光淬灭物理机制。稳态荧光发射光谱中,随着Fe3+浓度的增加,石墨烯氧化物的荧光强度急剧减弱。时间分辨荧光光谱和飞秒瞬态吸收光谱研究证实,加入不同浓度Fe3+的GO其动力学衰减曲线基本没有任何变化。结果表明,Fe3+对石墨烯氧化物的荧光淬灭主要是静态的荧光淬灭过程。Abstract: The graphene oxide(GO) is synthesized by the improved Hummer method. The fluorescence quenching mechanism of graphene oxide by Fe3+(0.5, 1, 2 mmol/L) in detail is studied using the time-resolved spectrometry probe system. From the steady photoluminescence emitting spectra, the fluorescence intensity of GO decreased dramatically as the Fe3+ concentration increased. From the time-resolved fluorescence spectra and femtosecond transient absorption spectra, the dynamic decay curves have no extinctive changes for GO with different Fe3+ concentrations. It is proved that the fluorescence quenching mechanism of GO by Fe3+ is mainly ascribed to the static fluorescence quenching.
-
Key words:
- graphene oxide /
- static fluorescence quenching /
- transient absorption /
- dynamic decay
-

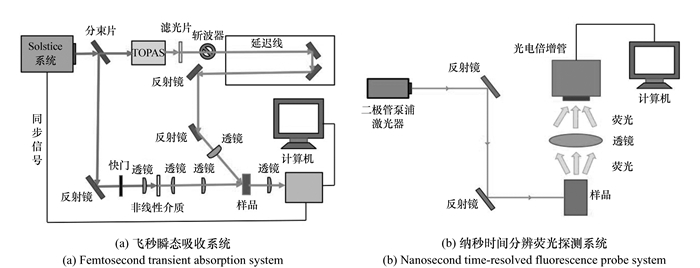
图 1 时间分辨光谱探测系统结构示意图
Figure 1. Schematic diagram of time-resolved spectroscopy probe system

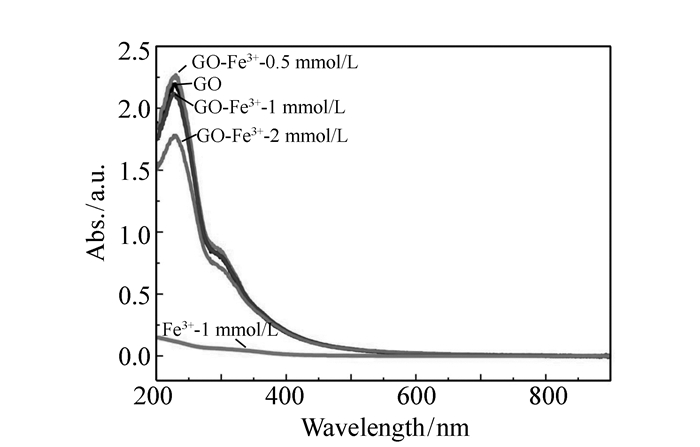
图 2 加入不同浓度Fe3+的GO的稳态吸收光谱
Figure 2. Steady-state absorption spectra of GO with different Fe3+ concentrations

图 3 加入不同浓度Fe3+的GO的荧光发射光谱
Figure 3. Steady-state emission spectra of GO with different Fe3+ concentrations

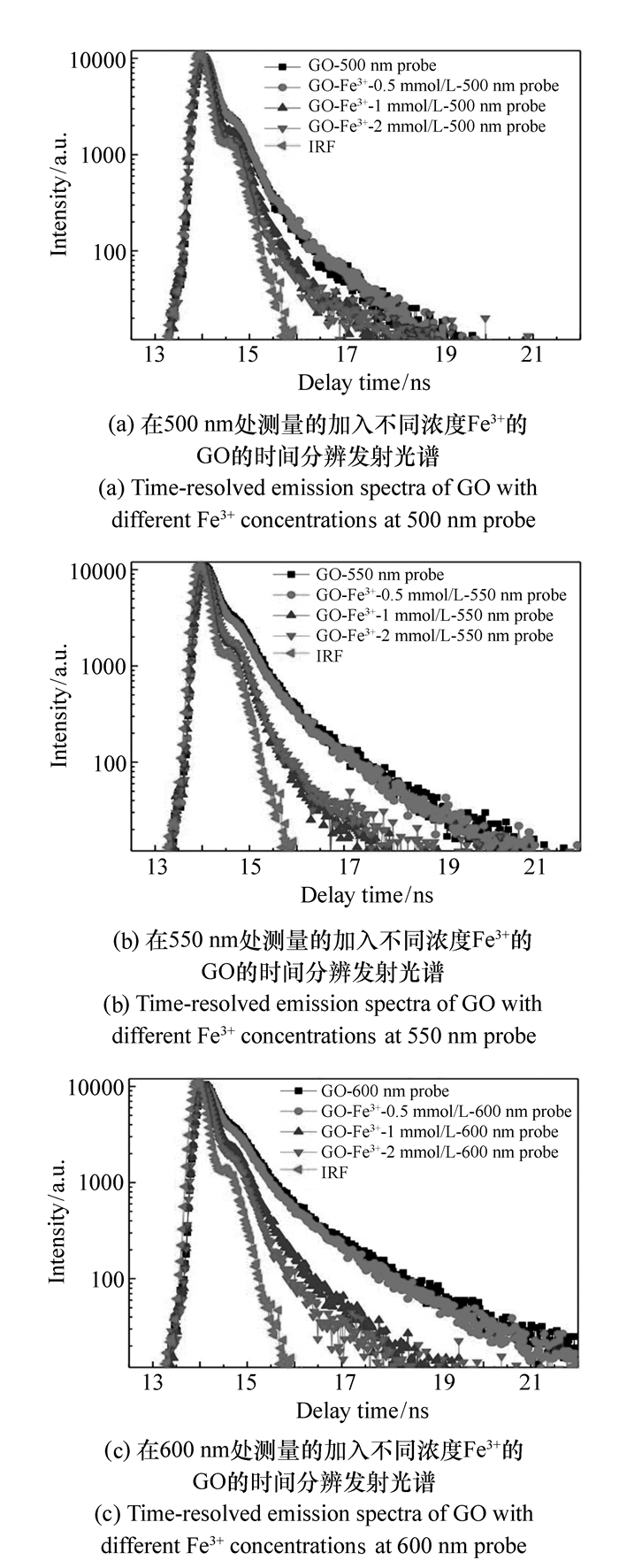
图 4 加入不同浓度Fe3+的GO的时间分辨发射光谱
Figure 4. $Time-resolved emission spectra of GO with different Fe3+ concentrations

图 5 加入不同浓度Fe3+的GO在400 nm激发下的瞬态吸收光谱
Figure 5. Transient absorption spectra of GO with different Fe3+ concentrations at 400 nm excitation

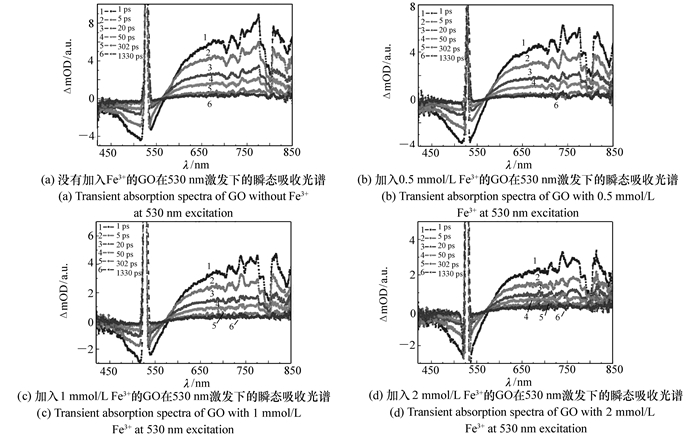
图 6 加入不同浓度Fe3+的GO在530 nm激发下的瞬态吸收光谱
Figure 6. Transient absorption spectra of GO with different Fe3+ concentrations at 530 nm excitation

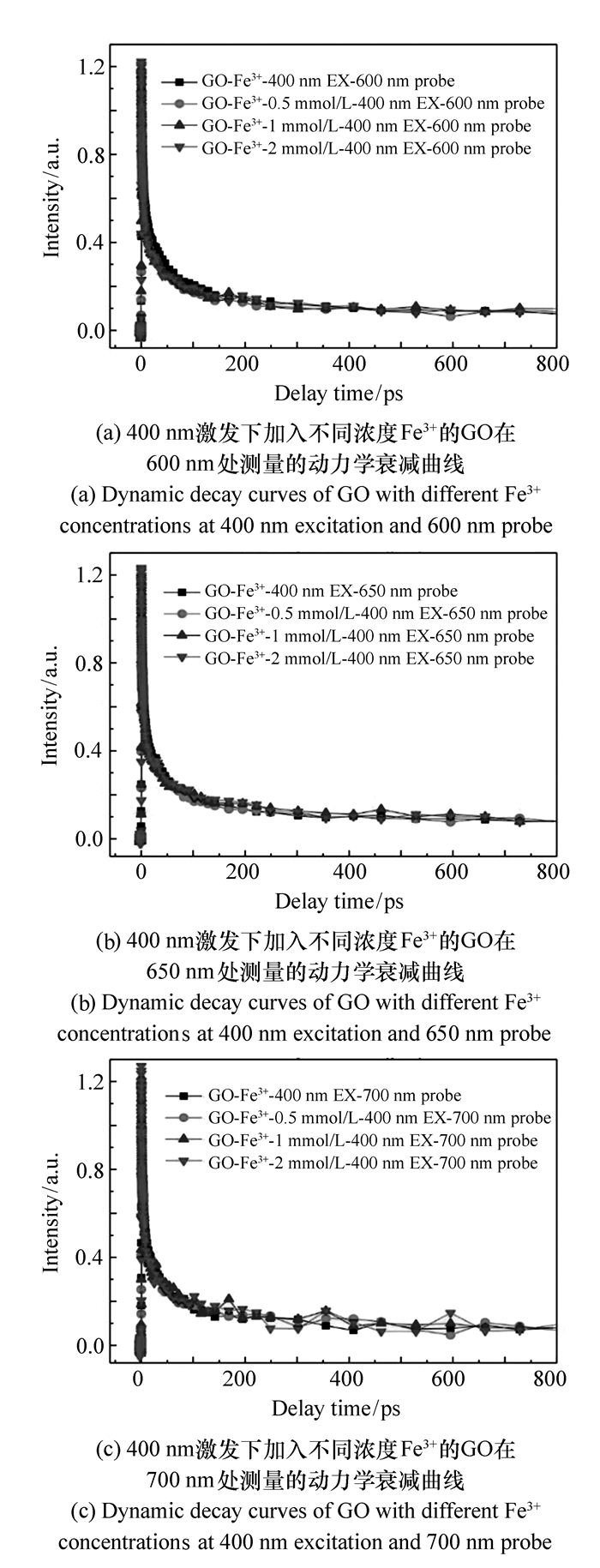
图 7 400 nm激发下加入不同浓度Fe3+的GO测量的动力学衰减曲线
Figure 7. Dynamic decay curves of GO with different Fe3+ concentrations at 400 nm excitation
-
[1] JOHARI P, SHENOY V B. Modulating optical properties of graphene oxide:role of prominent functional groups[J]. ACS Nano, 2011, 5:7640-7647. doi: 10.1021/nn202732t [2] LIU J, KIM G H, XUE Y, et al.. Graphene oxide nanoribbon as hole extraction layer to enhance efficiency and stability of polymer solar cells[J]. Adv. Mater., 2014, 26:786-790. doi: 10.1002/adma.201302987 [3] MCDONALD M P, ELTOM A, VIETMEYER F, et al.. Direct observation of spatially heterogeneous single-layer graphene oxide reduction kinetics[J]. Nano Lett., 2013, 13:5777-5784. doi: 10.1021/nl402057j [4] XIN G, MENG Y, MA Y, et al.. Tunable photoluminescence of graphene oxide from near-ultraviolet to blue[J]. Mater. Lett., 2012, 74:71-73. doi: 10.1016/j.matlet.2012.01.047 [5] MATHKAR A, TOZIER D, COX P, et al.. Controlled, stepwise reduction and band gap manipulation of graphene oxide[J]. J. Phys. Chem. Lett., 2012, 3:986-991. doi: 10.1021/jz300096t [6] LUO Z, VORA P M, MELE E J, et al.. Photoluminescence and band gap modulation in graphene oxide[J]. Appl. Phys. Lett., 2009, 94:111909. doi: 10.1063/1.3098358 [7] CHIEN C T, LI S S, LAI W J, et al.. Tunable photoluminescence from graphene oxide[J]. Angew. Chem. Int. Ed., 2012, 51:6662-6666. doi: 10.1002/anie.201200474 [8] THOMAS H R, VALL S C, YOUNG R J, et al.. Identifying the fluorescence of graphene oxide[J]. J. Phys. Chem. C, 2013, 1:338-342. http://pubs.rsc.org/en/content/articlelanding/2013/tc/c2tc00234e#! [9] LI M, CUSHING S K, ZHOU X, et al.. Fingerprinting photoluminescence of functional groups in graphene oxide[J]. J. Mater. Chem., 2012, 22:23374-23379. doi: 10.1039/c2jm35417a [10] ZHANG X F, SHAO X, LIU S. Dual fluorescence of graphene oxide:a time-resolved study[J]. J. Phys. Chem. A, 2012, 116:7308-7313. doi: 10.1021/jp301755b [11] KOZAWA D, MIYAUCHI Y, MOURI S, et al.. Exploring the origin of blue and ultraviolet fluorescence in graphene oxide[J]. J. Phys. Chem. Lett., 2013, 4:2035-2040. doi: 10.1021/jz400930f [12] CUSHING S K, LI M, HUANG F, et al.. Origin of strong excitation wavelength dependent fluorescence of graphene oxide[J]. ACS Nano, 2013, 8:1002-1013. http://www.ncbi.nlm.nih.gov/pubmed/24359152 [13] LIU Z, ROBINSON J T, SUN X, et al.. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs[J]. J. Am. Chem. Soc., 2008, 130:10876-10877. doi: 10.1021/ja803688x [14] CHEN J L, YAN X P. Ionic strength and pH reversible response of visible and near-infrared fluorescence of graphene oxide nanosheets for monitoring the extracellular pH[J]. Chem. Commun., 2011, 47:3135-3137. doi: 10.1039/c0cc03999c [15] SUN X, LIU Z, WELSHER K, et al.. Nano-graphene oxide for cellular imaging and drug delivery[J]. Nano Res., 2008, 1:203-212. doi: 10.1007/s12274-008-8021-8 [16] 李云飞, 陈洋, 毕宴钢, 等.还原GO-银纳米线柔性复合电极的制备与性能研究[J].发光学报, 2015, 36(5):545-551. doi: 10.3788/fgxbLI Y F, CHEN Y, BI Y G, et al.. Fabrication and characterization of reduced graphene oxide/silver nanowires flexible hybrid electrodes[J]. Chin. J. Lumin., 2015, 36(5):545-551.(in Chinese) doi: 10.3788/fgxb [17] 谢世伟, 肖啸, 谭建军, 等.基于石墨烯基电极染料敏化太阳能电池的研究进展[J].中国光学, 2014, 7(1):47-56. http://www.chineseoptics.net.cn/CN/abstract/abstract9095.shtmlXIE SH W, XIAO X, TAN J J, et al.. Recent progress in dye-sensitized solar cells using graphene-based electrodes[J]. Chinese Optics, 2014, 7(1):47-56.(in Chinese) http://www.chineseoptics.net.cn/CN/abstract/abstract9095.shtml [18] 董浩, 赵晓晖, 曲良东, 等.氧化石墨烯/硒化锌纳米光电材料的制备及其蓝光发射特性[J].发光学报, 2014, 7:767-771. http://www.cnki.com.cn/Article/CJFDTOTAL-FGXB201407003.htmDONG H, ZHAO X H, QU L D, et al.. Preparation and characteristics of reduced graphene oxide-zinc selenide nano optoelectronic materials[J]. Chin. J. Lumin., 2014, 7:767-771.(in Chinese) http://www.cnki.com.cn/Article/CJFDTOTAL-FGXB201407003.htm [19] WANG Y F, WANG H Y, LI Z S, et al.. Electron extraction dynamic decays in CdSe and CdSe/CdS/ZnS quantum dots adsorbed with methyl viologen[J]. J. Phys. Chem. C, 2014, 118:17240-17246. doi: 10.1021/jp5024789 [20] WANG L, ZHU S J, WANG H Y, et al.. Unraveling bright molecule-like state and dark intrinsic state in green-fluorescence graphene quantum dots via ultrafast spectroscopy[J]. Adv. Optical Mater, 2013, 1:264-271. doi: 10.1002/adom.201200020 [21] GAO B R, WANG H Y, SUN H B, et al.. Time-resolved fluorescence study of aggregation-induced emission enhancement by restriction of intramolecular charge transfer state[J]. J. Phys. Chem. B, 2010, 114:128-134. doi: 10.1021/jp909063d -
 下载:
下载:


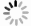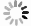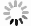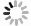
计量
- 文章访问数: 2607
- HTML全文浏览量: 594
- PDF下载量: 724
- 被引次数: 0
