


-
摘要: 为了拓展荧光辐射差分(Fluorescence Emission Difference,FED)显微术的应用,使得该方法可以同时对生物样品的不同组织结构进行超分辨成像,本文对双色FED显微系统展开了研究。FED的基本原理是将实心光斑扫描得到的共焦显微图像减去空心光斑扫描得到的负共焦图像,以此获得超分辨显微图像。在对单色FED显微系统进行研究后,本文提出了一种可行的双色FED显微成像系统方案。实验结果表明,在488 nm和640 nm激发光下,该系统在荧光颗粒上分别实现了135 nm和160 nm的空间分辨率,另外也能对生物样品的不同组织进行多色同时超分辨显微成像,满足了实际应用的要求。Abstract: To perform super-resolution imaging of different tissue structures of biological samples using fluorescence radiation differential microscopy simultaneously, a dual-color FED microscopy system is studied in this paper. The basic principle of the FED is to remove the confocal microscopy image obtained by scanning the solid spot from the confocal microscopy image obtained by scanning the hollow spot to obtain a super-resolution microscopy image. Based on the study of the monochromatic FED microscopy system, a feasible dual-color FED microscopy imaging system is proposed and imaging experiments are performed on fluorescent particles in this paper. The experimental results indicate that under excitation light of 488nm and 640nm, the system realizes spatial resolution of 135 nm and 160 nm of the fluorescent particles respectively. In addition, this system can also perform multi-color super-resolution microscopy imaging simultaneously on different tissues of biological samples, which meets the requirements of practical applications.
-

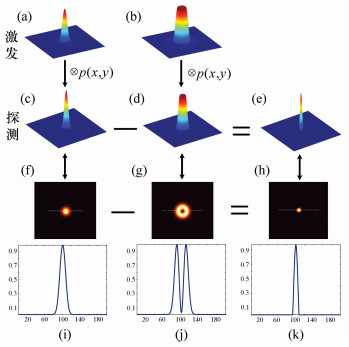
图 1 FED原理图解:(a),(b)分别为激发的实心光斑和空心光斑的PSF三维图;(c),(d)为探测的实心光斑和空心光斑的PSF三维图;(e)为FED的PSF三维图;(f),(g),(h)分别为(c),(d),(e)的平面图;(i),(j),(k)分别为(f),(g),(h)在虚线处的截面图
Figure 1. Illustration of FED theory. (a)PSF of confocal excited pattern; (b)PSF of negative confocal excited pattern; (c)PSF of confocal image; (d)PSF of negative confocal image; (e) PSF of FED image; (f), (g), (h) the plane graph of (c), (d) and (e); (i), (j), (k) the sectional drawing along the dash line of (f), (g) and (h)

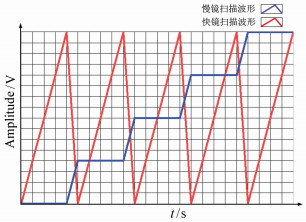
图 2 双色FED系统图与时序图(a)双色FED系统图,其中PBS为偏振分光镜,PM为涡旋位相板,RM为反射镜,DC为二色镜,4F SM为4F扫描模块,AL为消色差透镜,SMF为保偏光纤;(b)系统CAD设计图;(c)扫描时序图
Figure 2. Scheme and sequence chart of dual color FED system. (a)Dual color FED system, PBS:polarizing beam splitter, PM:vortex phase mask, RM:reflect mirror, DC:dichroic beam splitter, 4F SM:4F Scanning Module, AL:achromatic lens, SMF:single-mode polarization maintain fiber; (b)CAD of the system; (c)scan sequence chart

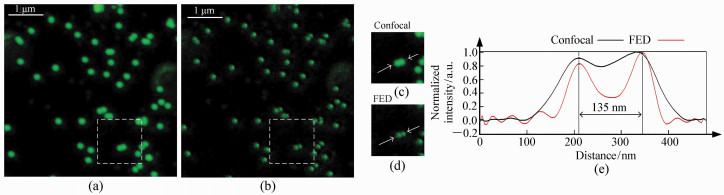
图 5 488 nm激发的荧光样品共焦和FED图像。(a)共焦图像;(b)FED图像;(c)共焦图像虚框内局部放大图像;(d)FED图像虚框内局部放大图像;(e)箭头部分归一化后的轮廓图
Figure 5. Confocal image and FED image of fluorescent beads excited at 488 nm. (a)Confocal image; (b)FED image. (c, d)Magnified view of dashed region at (a) and (b). (e)Normalized outline of arrow section of (c) and (d)

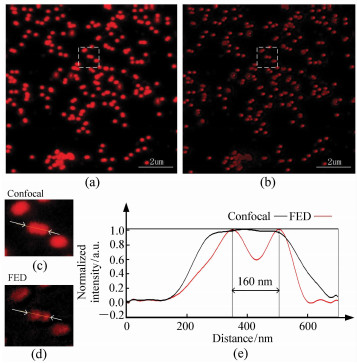
图 6 640 nm激发的荧光样品共焦和FED图像。(a)共焦图像;(b)FED图像;(c)共焦图像虚框内局部放大图像;(d)FED图像虚框内局部放大图像;(e)箭头部分归一化后轮廓图
Figure 6. Confocal image and FED image of fluorescent beads excited at 640 nm. (a)Confocal image; (b)FED image; (c, d)Magnified view of dashed region at (a) and (b); (e)Normalized outline of arrow section of (c) and (d)

图 7 生物样品多色成像图。(a)共焦图像;(b)FED图像;(c)共焦图像虚框内局部放大图;(d)FED图像虚框内局部放大图;(e)箭头指向部分轮廓图
Figure 7. Confocal image and FED image of biological structures excited at 488nm and 640nm simultaneously. (a)Confocal image; (b)FED image; (c, d)Magnified view of dashed region at (a) and (b); (e)Normalized outline of arrow section of (c) and (d)
-
[1] 李帅, 匡翠方, 丁志华, 等.受激发射损耗显微术(STED)的机理及进展研究[J].激光生物学报, 2013, 2:103-113. doi: 10.3969/j.issn.1007-7146.2013.02.002LI SH, KUANG C F, DING ZH H, et al.. A review on concept and development of stimulated emission depletion microscopy(STED)[J]. Acta Laser Biology Sinica, 2013, 2:103-113.(in Chinese) doi: 10.3969/j.issn.1007-7146.2013.02.002 [2] HUANG B, BATES M, ZHUANG X. Super-resolution fluorescence microscopy[J]. Annual Review of Biochemistry, 2009, 78:993-1016. doi: 10.1146/annurev.biochem.77.061906.092014 [3] HELL S W, WICHMANN J. Breaking the diffraction resolution limit by stimulated emission:stimulated-emission-depletion fluorescence microscopy[J]. Optics Letters, 1994, 19(11):780-782. doi: 10.1364/OL.19.000780 [4] KLAR T A, JAKOBS S, DYBA M, et al.. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission[J]. Proceedings of the National Academy of Sciences, 2000, 97(15):8206-8210. doi: 10.1073/pnas.97.15.8206 [5] KUANG C, LI S, LIU W, et al.. Breaking the diffraction barrier using fluorescence emission difference microscopy[J]. Scientific Reports, 2013, 3:1441. doi: 10.1038/srep01441 [6] 李帅. 超分辨荧光显微方法与系统研究[D]. 杭州: 浙江大学, 2014. http://cdmd.cnki.com.cn/Article/CDMD-10335-1014269194.htmLI SH. Research on super-resolution fluorescence microscopy method and system[D]. Huangzhou: Zhejiang University, 2014. (in Chinese) http://cdmd.cnki.com.cn/Article/CDMD-10335-1014269194.htm [7] MEYER L, WILDANGER D, MEDDA R, et al.. Dual-color STED microscopy at 30-nm focal-plane resolution[J]. Small, 2008, 4(8):1095-1100. doi: 10.1002/smll.v4:8 [8] LI S, KUANG C, HAO X, et al.. Enhancing the performance of fluorescence emission difference microscopy using beam modulation[J]. Journal of Optics, 2013, 15(12):125708. doi: 10.1088/2040-8978/15/12/125708 [9] YOU S, KUANG C, RONG Z, et al.. Eliminating deformations in fluorescence emission difference microscopy[J]. Optics Express, 2014, 22(21):26375-26385. doi: 10.1364/OE.22.026375 [10] RONG Z, LI S, KUANG C, et al.. Real-time super-resolution imaging by high-speed fluorescence emission difference microscopy[J]. Journal of Modern Optics, 2014, 61(16):1364-1371. doi: 10.1080/09500340.2014.933272 [11] HAO X, KUANG C F, WANG T T, et al.. Effects of polarization on the de-excitation dark focal spot in STED microscopy[J]. Journal of Optics, 010, 12(11):115707. http://cn.bing.com/academic/profile?id=076fc5db3c528172e3bafe865a738aff&encoded=0&v=paper_preview&mkt=zh-cn [12] WILSON T. Resolution and optical sectioning in the confocal microscope[J]. Journal of Microscopy, 2011, 244(2):113-121. doi: 10.1111/jmi.2011.244.issue-2 [13] BINGEN P, REUSS M, ENGELHARDT J, et al.. Parallelized STED fluorescence nanoscopy[J]. Optics Express, 2011, 19(24):23716-23726. doi: 10.1364/OE.19.023716 [14] RANKIN B R, KELLNER R R, HELL S W. Stimulated-emission-depletion microscopy with a multicolor stimulated-Raman-scattering light source[J]. Optics Letters, 2008, 33(21):2491-2493. doi: 10.1364/OL.33.002491 [15] STAUDT T M. Strategies to reduce photobleaching, dark state transitions and phototoxicity in subdiffraction optical microscopy[D]. Ruperto-Carola University of Heidelberg, Germany, 2009. https://www.researchgate.net/publication/33429512_Strategies_to_reduce_photobleaching_dark_state_transitions_and_phototoxicity_in_subdiffraction_optical_microscopy [16] HAO X, ALLGEYER E S, BOOTH M J, et al.. Point-spread function optimization in isoSTED nanoscopy[J]. Optics Letters, 2015, 40(15):3627-3630. doi: 10.1364/OL.40.003627 [17] HIGDON P D, T R K P, WILSON T. Imaging properties of high aperture multiphoton fluorescence scanning optical microscopes[J]. Journal of Microscopy, 1999, 193(2):127-141. doi: 10.1046/j.1365-2818.1999.00448.x [18] RUST M J, BATES M, ZHUANG X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy(STORM)[J]. Nature Methods, 2006, 3(10):793. doi: 10.1038/nmeth929 [19] BRODEHL A, HEDDE P N, DIEDING M, et al.. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants[J]. Journal of Biological Chemistry, 2012, 287(19):16047-16057. doi: 10.1074/jbc.M111.313841 [20] CARRINGTON W A, LYNCH R M, MOORE E D, et al.. Superresolution three-dimensional images of fluorescence in cells with minimal light exposure[J]. Science, 1995, 268(5216):1483-1487. doi: 10.1126/science.7770772 -
 下载:
下载:



计量
- 文章访问数: 2986
- HTML全文浏览量: 1082
- PDF下载量: 199
- 被引次数: 0
